Chapter 8 Statistical significance and p-values
Frequentist statistics works by asking what would have happened if we were to repeat a data collection exercise many times, assuming that the population remains the same each time. This is the basic idea we used to generate sampling distributions in the last chapter. The details of this procedure depend on what kind of question we are asking, which varies from one situation to another.
What is common to every frequentist technique is that we ultimately have to work out what some kind of sampling distribution looks like. Once we’ve done that we can evaluate how likely a particular result is. This naturally leads onto the most important ideas in frequentist statistics: p-values and statistical significance.
8.1 Estimating a sampling distribution
Let’s carry on with the plant polymorphism example. Our ultimate goal is to find out if the purple morph frequency is likely to be greater than 25% in the new study population. The suggestion above is that we will need to work out what the sampling distribution of the purple morph frequency estimate looks like to get to this point.
At first glance this seems an impossible task in the real world because we would only have access to a single sample? The solution to this problem is surprisingly simple: use the one sample to approximate the population in some way, then work out what the sampling distribution of our estimate should look like by ‘taking samples’ from this approximation.
We’ll unpack this idea a bit more before we try it out for real.
8.1.1 Overview of bootstrapping
There are many ways to use a sample to approximate the population it came from. One of the simplest is to pretend the sample is the true population. All we then have to do to get at a sampling distribution is draw new samples from this pretend population. This may sound like ‘cheating’ but it turns out this is a perfectly valid way to construct approximate sampling distributions.
We’ll try to get a sense of how this works using a physical analogy based on our plant morph example. Imagine that we have written down the colour of every sampled plant on a different piece of paper and then placed these bits of paper into in a hat. We then do the following:
- Pick a piece of paper at random, record its value (purple or green), put the paper back into the hat, and shake the hat about to mix up the bits of paper.
(The shaking here is meant to ensure that each piece of paper has an equal chance of being picked.)
Now pick another piece of paper (we might get the same one), record its value, put that one back into the hat, and shake everything up again.
Repeat this process until we have a recorded new sample of colours that is the same size as the real sample. We have now have generated a ‘new sample’ from the original one.
(This process is called ‘sampling with replacement.’ Each artificial sample is called a ‘bootstrapped sample.’)
For each bootstrapped sample, calculate whatever quantity is of interest. In our example, this is the proportion of purple plants sampled.
Repeat steps 1-4 until we have generated a large number of bootstrapped samples. About 10000 is sufficient for most problems.
Although it seems like cheating this procedure really does approximate the sampling distribution of the purple plant frequency. It is called bootstrapping (or ‘the bootstrap’).
The bootstrap is quite a sophisticated technique developed by statistician Bradley Efron. We’re not going to use it to solve real data analysis problems and there’s no need to learn how to do it. We’re introducing bootstrapping because it provides a reasonably intuitive way to understand how frequentist methodology works without having to get stuck into any difficult mathematics.
8.1.2 Doing it for real
No one carries out bootstrapping using bits of paper and hat. Generating 10000 bootstrapped samples via such a method would obviously take a very long time! Luckily, computers are very good at carrying out repetitive tasks quickly. We’re going to work through how to implement the bootstrap for our hypothetical example.
The best way to understand what follows is to actually work through the example. You are strongly encouraged to do this!
Set up and read the data
Assume we had sampled 250 individuals from the new plant population. A data set representing this situation is stored in the Comma Separated Value (CSV) file called ‘MORPH_DATA.CSV.’ Using the template project for the book examples, run through the following steps in your script:
Read the data into an R data frame, assigning it the name
morph_data.Use functions like
glimpse/strto inspectmorph_data.Use the
Viewfunction to inspect the data with RStudio.
Make sure you can answer the following questions:
How many observations are in the data set?
How many variables are in the data set? What are their names?
What kind of variables are they?
What values do the different variables take?
The point of all this is to check that we understand our data. Always examine a dataset after reading it into R. If we don’t understand how our data is organised and what variables we are working with we are bound to make otherwise avoidable mistakes.
What you should have found is that morph_data contains 250 rows and two columns/variables: Colour and Weight. Colour is a categorical variable and Weight is a numeric variable. The Colour variable contains the colour of each plant in the sample.
What is that Weight variable all about? Actually… we don’t need this now—we’ll use it in the next chapter.
Running the bootstrap
Now that we understand the data we’re ready to implement bootstrapping. We are going to use a few R programming tricks that you may not have come across before. We’ll explain these as we go but there’s really no need to learn them. Focus on the ‘why’—the logic of what we’re doing—rather than the ‘how.’
We want to construct an approximate sampling distribution for the frequency of purple morphs. That means the variable that matters is Colour. Rather than work with this inside the morph_data data frame, we’re going to pull it out using the $ operator and assign it a name (plant_morphs):
# pull out the 'Colour' variable
plant_morphs <- morph_data$Colour Next, we’ll take a quick look at the values of plant_morphs:
# what is the set of values 'plant_morphs' can take?
unique(plant_morphs)## [1] "Green" "Purple"# show the first 100 values
head(plant_morphs, 50) ## [1] "Green" "Green" "Green" "Purple" "Green" "Green" "Green" "Green"
## [9] "Green" "Green" "Green" "Green" "Green" "Purple" "Green" "Green"
## [17] "Purple" "Purple" "Green" "Green" "Green" "Green" "Green" "Purple"
## [25] "Green" "Green" "Green" "Green" "Purple" "Purple" "Green" "Green"
## [33] "Green" "Purple" "Purple" "Green" "Green" "Green" "Green" "Purple"
## [41] "Green" "Purple" "Green" "Green" "Purple" "Purple" "Green" "Green"
## [49] "Green" "Green"The last line printed out the first 50 values of plant_morphs. This shows that plant_morphs is a simple character vector with two categories describing the plant colour morph information.
Next, we calculate and store the sample size (samp_size) and the point estimate of purple morph frequency (mean_point_est) from the sample:
# get the sample size form the length of 'plant_morphs'
samp_size <- length(plant_morphs)
samp_size## [1] 250# estimate the frequency of purple plants as a %
mean_point_est <- 100 * sum(plant_morphs == "Purple") / samp_size
mean_point_est## [1] 30.8The code in the point estimate calculation says “add up all the cases where plant_morphs is equal to ‘purple,’ divide that total by the sample size to get the proportion of purple plants in the sample, then multiply th eproprtion by 100 to turn it into a percentage.” So… we find that 30.8% of plants were purple among our sample of 250 plants.
Now we’re ready to start bootstrapping. For convenience, we’ll store the number of bootstrapped samples we want in n_samp (i.e. 10000 in this case):
# number of bootstrapped samples we want
n_samp <- 10000Next we need to work out how to resample the values in the plant_morphs vector. The sample function can do this for us:
# resample the plant colours
samp <- sample(plant_morphs, replace = TRUE)
# show the first 50 values of the bootstrapped sample
head(samp, 50) ## [1] "Purple" "Green" "Green" "Green" "Green" "Purple" "Purple" "Green"
## [9] "Green" "Purple" "Purple" "Green" "Green" "Green" "Green" "Green"
## [17] "Green" "Green" "Green" "Purple" "Purple" "Purple" "Purple" "Green"
## [25] "Green" "Green" "Purple" "Green" "Green" "Purple" "Green" "Purple"
## [33] "Green" "Green" "Green" "Purple" "Purple" "Green" "Purple" "Green"
## [41] "Purple" "Purple" "Purple" "Green" "Green" "Purple" "Green" "Green"
## [49] "Purple" "Green"The replace = TRUE ensures that we sample with replacement—this is the ‘putting the bits of paper back in the hat’ part of the process.
The new samp variable now contains exactly one bootstrapped sample of th 250 plants in the real sample. We only need to extract one number from this—the frequency of purple morphs:
# calculate the purple morph frequencyin the bootstrapped sample
first_bs_freq <- 100 * sum(samp == "Purple") / samp_sizeThat’s one bootstrapped value of the purple morph frequency. Fine, but we need \(10^{4}\) values. We don’t want to have to keep doing this over an over ‘by hand’—making second_bs_freq, third_bs_freq, and so on—because this would be very slow and boring to do.
As we said earlier, computers are very good at carrying out repetitive tasks. The replicate function can replicate any R code many times and return the set of results. Here is some R code that repeats what we just did n_samp times, storing the resulting bootstrapped values of purple morph frequency in a numeric vector called boot_out:
boot_out <- replicate(n_samp, {
samp <- sample(plant_morphs, replace = TRUE)
100 * sum(samp == "Purple") / samp_size
})The boot_out vector now contains a bootstrapped sample of frequency estimates. Here are the first 30 values rounded to 1 decimal place:
head(boot_out, 30) %>% round(1)## [1] 31.6 24.0 26.8 29.2 32.8 26.4 34.4 27.6 33.2 32.8 33.6 26.0 34.0 29.6 29.6
## [16] 31.2 29.6 30.4 29.6 27.2 30.8 28.8 29.6 30.4 32.8 30.0 31.6 35.6 34.4 32.0(We used the pipe %>% to make a code a bit more readable—remember, this won’t work unless the dplyr package is loaded.)
Making sense of the bootstrapped sample
What has all this achieved? The numbers in boot_out represent the values of purple morph frequency we can expect to find if we repeated the data collection exercise many times, under the assumption that the purple morph frequency is equal to that of the actual sample. This is a bootstrapped sampling distribution!
We can use this bootstrapped sampling distribution in a number of ways. Let’s plot it first get a sense of what it looks like. A histogram is OK here because we have a reasonably large number of possible cases:
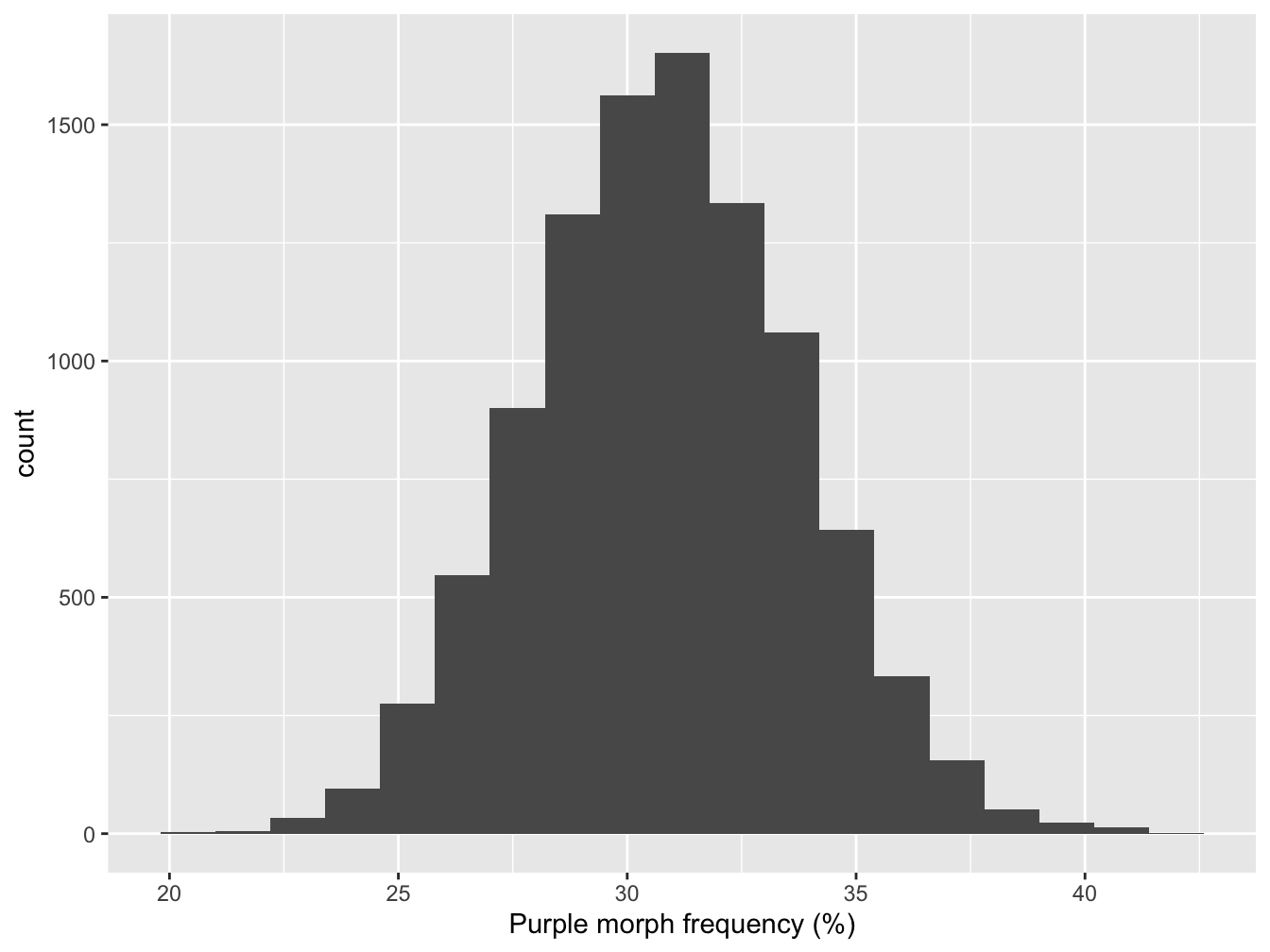
Figure 8.1: Bootstrapped sampling distribution of purple morph frequency
What the most common values in our bootstrapped sample? The centre of the distribution looks to be round about 30%. We can be a bit more precise by calculating its mean:
mean(boot_out) %>% round(1)## [1] 30.8This is essentially the same as the point estimate of purple morph frequency from the true sample. In fact, it is guaranteed to be the same if we construct a large enough sample because we’re just resampling the data used to estimate that frequency.
A more useful quantity is the standard error (SE) of our estimate. This is defined as being the standard deviation of the sampling distribution. We can calculate that by applying the sd function to the bootstrapped sample:
sd(boot_out) %>% round(1)## [1] 2.9The standard error is a very useful quantity. Remember, it is a measure of the precision of an estimate. For example, a large SE implies that our sample size was too small to reliably estimate the population mean; a small SE means we have a reliable estimate. Once we have the point estimate of a population parameter its standard error we’re able to start asking questions like, “is the true value likely to be different from 25%.”
It is standard practice include the standard error when we report a point estimate of some quantity. Whenever we report a point estimate, we really should also report its standard error, like this:
The frequency of purple morph plants (n = 250) was 30.8% (s.e. ± 2.9).
Notice we also report the sample size. More on that later in the book.
8.2 Statistical significance
Now back to the question that motivated all the work in the last few chapters. Is the purple morph frequency greater than 25% in the new study population? The first thing to realise is that we can never answer a question like this definitively from a sample. We have to carry out some kind of probabilistic assessment instead. To make this assessment, we’re going to do something that looks odd at first glance.
Don’t panic! This stuff is hard.
The ideas in this next section are very abstract and you may not understand them straight away. That’s fine. Don’t worry—these ideas take time to absorb and understand.
Carrying out the assessment
We need to make two assumptions to arrive at our probabilistic assessment of whether or not the purple morph frequency greater than 25%:
Assume the true value of the purple morph frequency in our new study population is 25%, i.e. we’ll assume the population parameter of interest is the same as that of the original population that motivated this work. In effect, we’re pretending there is really no difference between the populations.
Assume that the form of sampling distribution that we just generated would have been the same if the ‘equal population’ hypothesis were true. That is, the expected ‘shape’ of the sampling distribution would not change if the purple morph frequency really was 25%.
That first assumption is an example of a null hypothesis. The null hypothesis is an hypothesis of ‘no effect’ or ‘no difference.’ We’re going to revisit this idea many times in future chapters.
The second assumption is necessary for the reasoning below to work. In fact, this can be shown to be a pretty reasonable assumption in many situations. We don’t want to get lost in the details though so you will have to trust us on this one.
Now we ask a question: if the purple morph frequency in the population really is 25%, what would its corresponding sampling distribution look like? This is called the null distribution—the distribution expected under the null hypothesis.
If the second assumption is valid, we can actually construct the null distribution from our bootstrapped distribution as follows:
null_dist <- boot_out - mean(boot_out) + 25All we did here was shift the bootstrapped sampling distribution along until the mean is at 25%. Here’s what that null distribution looks like, along with the original observed estimate of the purple morph frequency:
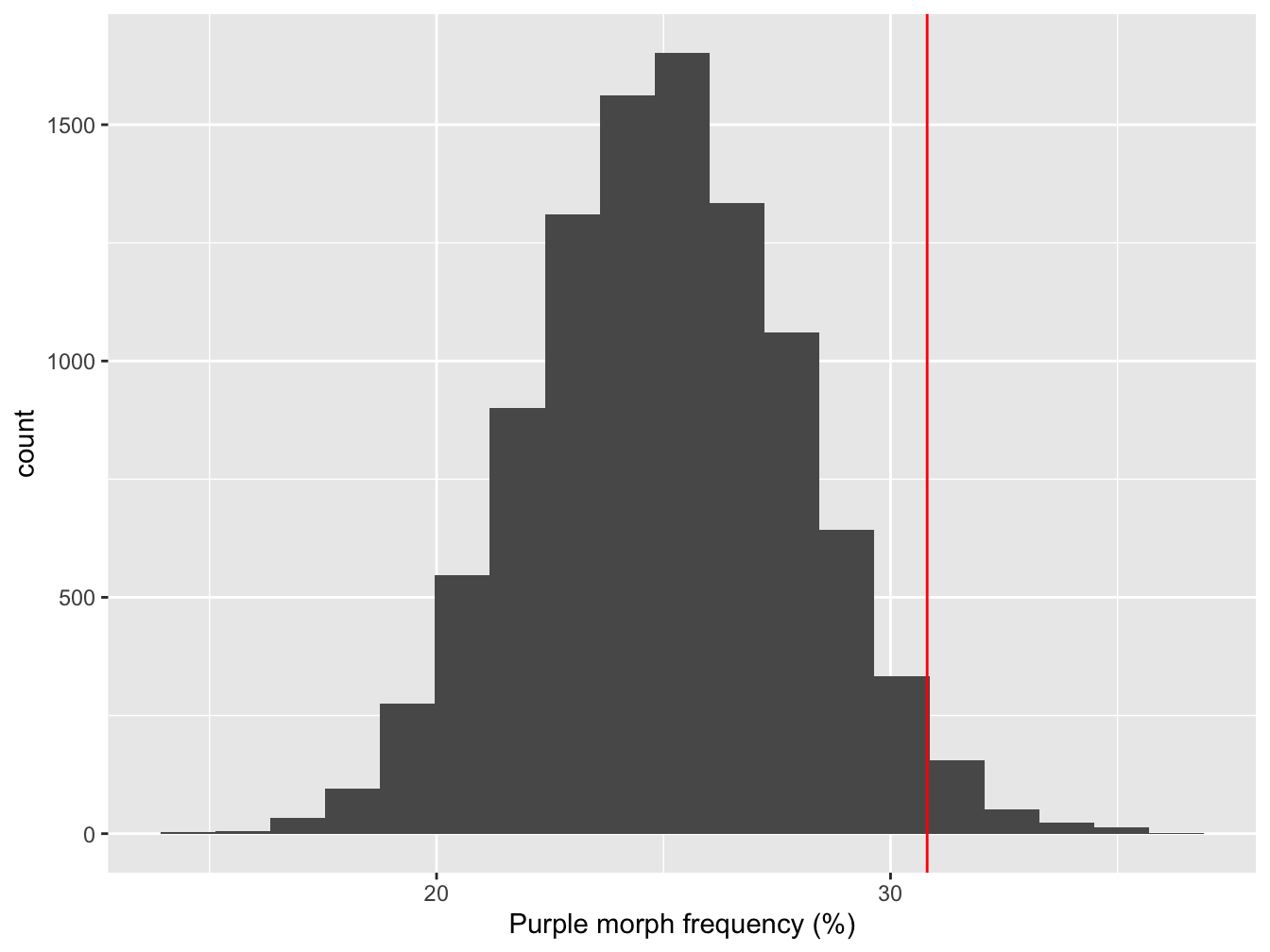
Figure 8.2: Sampling distribution of purple morph frequency under the null hypothesis
The red line shows where the point estimate from the true sample lies. What does this tells us? It looks like the observed purple morph frequency would be quite unlikely to have arisen through sampling variation if the population frequency really was 25%. We can say this because the observed frequency (red line) lies at the end of one ‘tail’ of the sampling distribution over on the right.
We need to be able to make a more precise statement than this though. Instead of ‘eyeballing’ the distribution, we can quantify how often the values of the bootstrapped null distribution ended up greater than the observed estimate:
p_value <- sum(null_dist > mean_point_est) / n_samp
p_value## [1] 0.0246This number (generally denoted ‘p’) is called a p-value.
Interpreting the p-value
What are we supposed to do with the finding p = 0.0246? This is the probability of obtaining a result equal to, or ‘more extreme,’ than that which was actually observed, assuming that the hypothesis under consideration (the null hypothesis) is true. The null hypothesis is one of no effect (or no difference), and so a low p-value can be interpreted as evidence for an effect being present. It’s worth reading that a few times…
In our example, it appears that the purple morph frequency we observed is fairly unlikely to occur if its frequency in the new population really was 25%. In biological terms, we take the low p-value as evidence for a difference in purple morph frequency among the populations, i.e. the data supports the prediction that the purple morph is present at a frequency greater than 25% in the new study population.
One important question remains: How small does a p-value have to be before we are happy to conclude that the effect we’re interested in is probably present? In practice, we do this by applying a threshold, called a significance level. If the p-value is less than the chosen significance level we say the result is said to be statistically significant. Most often (in biology at least), we use a significance level of p < 0.05 (5%).
Why do we use a significance level of p < 0.05? The short answer is that this is just a convention. Nothing more. There is nothing special about the 5% threshold, other than the fact that it’s the one most often used. Statistical significance has nothing to do with biological significance. Unfortunately, many people are very uncritical about the use of this arbitrary threshold, to the extent that it can be very hard to publish a scientific study if it doesn’t contain ‘statistically significant’ results.
8.3 Concluding remarks
We just carried out a type of statistical test called a significance test. It was a bit convoluted reasoning, but the chain of reasoning we just employed underlies all the significance tests we use in this book. The precise details of how to construct such tests will vary from one problem to the next, but ultimately, when using frequentist ideas we always…
assume that there is actually no ‘effect’ (the null hypothesis), where an effect is expressed in terms of one or more population parameters,
construct the corresponding null distribution of the estimated parameter by working out what would happen if we were to take frequent samples in the ‘no effect’ situation,
(This is why the word ‘frequentist’ is used to describe this flavour of statistics.)
- then compare the estimated population parameter to the null distribution to arrive at a p-value, which evaluates how frequently the result, or a more extreme result, would be observed under the hypothesis of no effect.
We used the bootstrap to operationalise that process for our example. Bootstrapping is certainly a useful tool but it is also quite an advanced technique that can be difficult to apply in many settings. We won’t use it any more—the bootstrap was introduced here to demonstrate how frequentist reasoning works.
We will focus on simple, ‘off-the-shelf’ statistical tools in this book. The good news is we don’t need to understand the low-level details to use these tools effectively. As long as we’re able to identify the null hypothesis and understand how to interpret the associated p-values we should be in a good position to apply them. These two ideas—null hypotheses and p-values—are so important, we’re going consider them in much greater detail over the next two chapters.